
A peer-reviewed electronic journal published by the
Institute for Ethics and ISSN 1541-0099 |
|
The intravenous median lethal dose (LD50) for 50% of hosts inoculated with various bacteremic microorganisms ranges widely from 1-109 CFU/gm (Table 2), but the central range appears to be 0.1-100 ×106 CFU/ml assuming a ~1 gm/cm3 density for biological materials.
2.2 ViremiaViremia is the presence of virus particles in the bloodstream, usually a transient condition [7]. Viruses are acellular bioactive parasites that attack virtually every form of cellular life. Viruses have diameters ranging from 16-300 nm [52] -- for example, poliomyelitis ~18 nm, yellow fever ~25 nm, adenovirus (common cold) ~70 nm, influenza (flu) ~100 nm, herpes simplex and rabies ~125 nm, and psittacosis ~275 nm [53]. Their shape is either pseudospherical with icosahedral symmetry, as in the poliomyelitis virus, or rodlike, as in the tobacco mosaic virus (TMV). A virus surrounded only by protein coat (capsid) is a naked virus; some viruses (e.g., HIV, HSV, pox), called enveloped viruses, acquire a lipid membrane envelope from their host cell upon release.In cases of blood plasma viremia, virion particle
counts range from 1/ml to 0.35 ×106/ml for HIV in humans
[54-56],
with a mean of 25/ml for asymptomatic patients; viral loads for
simian immunodeficiency virus (SIV) in monkeys may be much higher,
2-200 ×106/ml of blood [57].
Hepatitis C (HCV) [58]
infectious viral loads (at 2.3 FungemiaIn severely immunocompromised patients, fungi may gain access to the bloodstream, producing fungemia [7]. Fungal cells in peripheral blood are typically ovoid to elongated, from 3 × 3 microns up to 7 ×10 microns in size, and occur singly, budding, or in short chains and clusters [61]. Candidal fungemia is most common; Candida albicans blood counts in human patients are considered "ultralow" at < 1 CFU/ml and "low" at 1-3 CFU/ml in neonates [62], but "high" at > 5 CFU/ml in adult patients [63]; in one test series, fungemic patients showed 5.5 CFU/ml in venous blood and 9.1 CFU/ml in arterial blood, suggesting that peripheral tissues may clear ~40% of yeasts [64]. Rats injected with ~100 ×106 CFU/ml of C. albicans all died in < ~6 hours from nonendotoxemic (i.e., non-LPS related) shock [65].Patients with catheter-related fungemia due to fungus counts of Malassezia furfur at 50-1000 CFU/ml required antibiotic treatment [66], and catheter-related Rhodotorula (red yeast) infected patients with colony counts in the 100-1000 CFU/ml range required antifungal therapy [67]. Human bloodstream fungal infections thus appear to range from 1-1000 CFU/ml. Disseminated (systemic) candidiasis is effectively managed with 0.2 gm/day of fluconazole for at least 4 weeks [12]. Coccidioides immitis fungal infection is treated with ~0.02 gm/day (~200 ml/day I.V. drip solution via Ommaya reservoir into the brain ventricles) of amphotericin B for up to 9-11 months [12] (very toxic, with overdose leading to cardio-respiratory arrest; typically dosed as total cumulative). Respiratory fungal histoplasmosis (Histoplasma capulatum) may be treated with oral doses of itraconazole at 0.2-0.5 gm/day for a minimum of 3 months [12]. 2.4 Parasitemia and RickettsemiaParasitemia arises from parasites that have evolved to live in the bloodstream include the Plasmodium (malaria) family and the flagellate protozoans Trypanosoma (sleeping sickness) and Leishmania (leishmaniasis). Blood parasites typically have a juvenile form that is ovoid or ring-shaped with dimensions of 1-5 microns, and an adult tubular form measuring 1-5 microns in width and 10-30 microns in length [68]. In Trypanosoma brucei, the number of trypanosomes in blood fluctuates in waves, and the organisms are typically undetectable for 3 out of 5 days [69]. Trypomastigotes have an I.V. LD50 in mice of ~2.5/gm [70, 71]. Trypanosoma brucei gambiense inoculated into mice has an LD50 of 0.02-0.15 ×106 trypanosomes/gm, with growth rates slowing at organism blood concentrations > 300 ×106 trypanosomes/ml and death occurring at a blood parasite load of 2000 ×106 trypanosomes/ml [72]. Malaria may be treated with several oral doses of chloroquine phosphate totalling 2.5 gm over three days, but there is increasing microbial resistance to chloroquine worldwide and as little as 1 gm of the medicine can be fatal in children, with toxic symptoms appearing within minutes of overdosage [12]; a single 1.25 gm dose of mefloquine is sometimes effective in mild cases [12].Rickettsia are
rod-shaped or coccoid gram-negative obligate intracellular parasites
~0.25 microns in diameter that in humans grow principally in
endothelial cells of small blood vessels, producing vasculitis, cell
necrosis, vessel thrombosis, skin rashes and organ dysfunctions [73].
The infection is characterized by repetitive cycles of bloodborne
organisms, or rickettsemia. For example, in cattle the number of
pathogens in the blood varies between a low of 100/ml and a peak of
1-10 ×106/ml over 6-8 week intervals; in each cycle, the
blood count slowly rises over 10-14 days and then declines
precipitously [74].
However, most of these parasites are found in the red cells, and the
organism's appearance in the blood plasma is incidental to its
activity. Plasma titers for free R. rickettsii organisms in
the blood of human patients with Rocky Mountain spotted fever
averaged 5-16 parasites/ml in treated patients who survived, and
1000 parasites/ml in the postmortem plasma of one patient with
untreated fatal fulminant fever [75].
Antibiotic therapy has reduced the death rate from 20% to about 7%,
with death usually occurring when treatment is delayed [8].
3. Microbivore Scaling Analysis and Baseline DesignThe foregoing review suggests that existing treatments for many septicemic agents often require large quantities of medications that must be applied over long periods of time, and often achieve only incomplete eradication, or merely growth arrest, of the pathogen. A nanorobotic device that could safely provide quick and complete eradication of bloodborne pathogens using relatively low doses of devices would be a welcome addition to the physician's therapeutic armamentarium. The following analysis assumes a bacterial target (e.g. bacteremia), although other targets are readily substituted (Section 4.4).The microbivore is an oblate spheroidal nanomedical device consisting of 610 billion precisely arranged structural atoms plus another 150 billion mostly gas or water molecules when fully loaded (Section 3.2.5). The nanorobot measures 3.4 microns in diameter along its major axis and 2.0 microns in diameter along its minor axis, thus ensuring ready passage through even the narrowest of human capillaries (~4 microns in diameter [1, LINK]). Its gross geometric volume of 12.1056 micron3 includes two normally empty internal materials processing chambers totalling 4 micron3 in displaced volume. The device may consume up to 200 pW of continuous power while in operation and can completely digest trapped microbes at a maximum throughput of 2 micron3 per 30-second cycle, large enough to internalize almost all relevant microbes in a single gulp. As in previous designs [2], to help ensure high reliability the system presented here has tenfold redundancy in all major components, excluding only the largest passive structural elements. During each cycle of operation, the target bacterium is bound to the surface of the microbivore via species-specific reversible binding sites [1, LINK]. Telescoping robotic grapples emerge from silos in the device surface, establish secure anchorage to the microbe's plasma membrane, then transport the pathogen to the ingestion port at the front of the device where the cell is internalized into a morcellation chamber. After sufficient mechanical mincing, the morcellated remains are pistoned into a digestion chamber where a preprogrammed sequence of engineered enzymes are successively injected and extracted, reducing the morcellate primarily to monoresidue amino acids, mononucleotides, glycerol, free fatty acids and simple sugars, which are then harmlessly discharged into the environment through an exhaust port at the rear of the device, completing the cycle. This "digest and discharge" protocol [1, LINK, LINK] is conceptually similar to the internalization and digestion process practiced by natural phagocytes, but the artificial process should be much faster and cleaner. For example, it is well-known that macrophages release biologically active compounds such as muramyl peptides during bacteriophagy [76], whereas well-designed microbivores need only release biologically inactive effluent. 3.1 Primary Phagocytic SystemsThe principal activity which drives microbivore scaling and design is the process of digestion of organic substances, which also has some similarity to the digestion of food. The microbivore digestive system has four fundamental components -- an array of reversible binding sites to initially bind and trap target microbes (Section 3.1.1), an array of telescoping grapples to manipulate the microbe, once trapped (Section 3.1.2), a morcellation chamber in which the microbe is minced into small, easily digested pieces (Section 3.1.3), and a digestion chamber where the small pieces are chemically digested (Section 3.1.4).3.1.1 Reversible Microbial Binding SitesThe first function the microbivore must perform is to acquire a pathogen to be digested. A collision between a bacterium of the target species and the nanorobotic device brings their surfaces into intimate contact, allowing reversible binding sites on the microbivore hull to recognize and weakly bind to the bacterium. Binding sites can already be engineered [77, 78]. Bacterial membranes are quite distinctive, including such obvious markers as the family of outer-membrane trimeric channel proteins called porins in gram-negative bacteria like E. coli [79, 80] and other surface proteins such as Staphylococcal protein A [81] or endotoxin (lipopolysaccharide or LPS), a variable-size carbohydrate chain that is the major antigen of the outer membrane of gram-negative bacteria. Mycobacteria contain mycolic acid in their cell walls [82]. And only bacteria employ right-handed amino acids in their cellular coats, which helps them resist attack by digestive enzymes in the stomach and by other organisms. Peptidoglycans, the main structural component of bacterial walls, are cross-linked with peptide bridges that contain several unusual nonprotein amino acids and D-enantiomeric forms of Ala, Glu, and Asp [83]. D-alanine is the most abundant D-amino acid found in most peptidoglycans and the only one that is universally incorporated [84]. Macrophages have evolved a variety of plasma membrane receptors that recognize conserved motifs having essential biological roles for pathogens, hence the surface motifs are not subject to high mutation rates; these pathogen receptors on macrophages have been called "pattern recognition receptors" and their targets "pathogen-associated molecular paterns" [246]. Genomic differences between virulent and non-pathogenic bacterial strains [85] likely produce phenotypic differences that could enable the biasing of nanorobots towards the detection of the more toxic variants, if necessary.Additionally, all bacteria of a given species
express numerous unique proteins in their outermost coat. A complete
review is beyond the scope of this paper, but a few representative
examples can be cited. Each single-celled Staphylococcus aureus
organism displays binding sites for human vitronectin on its
surface, including 260 copies/cell representing high-affinity sites
and 5,240 copies/cell representing moderate-affinity sites [86].
The plasmid-specified major outer membrane protein TraTp of
Escherichia coli is normally present in 21,000 copies/cell at
the cell surface [87].
Streptococcus pyogenes (strain 6414) has 11,600 copies/cell
of surface binding sites to human collagen [88];
another receptor protein specific to type II collagen (among the
dozens of collagen types) are found in 30,000 copies/cell on the
surface of each Staphylococcus aureus (strain Cowan 1) cell
with equilibrium constant Kd
= Assuming that nine species-specific bacterial coat ligands are sufficient to uniquely identify an encountered bacterium as belonging to the target species or strain, and that ~104 copies of each of the nine ligands are present on a bacterial surface of area ~10 micron2, then the mean distance between each ligand of the same type is 31.6 nm. A square array of 200 adjacent ligand receptors on the nanorobot surface, with each ligand or receptor active site ~5 nm2 in area (e.g., antibody-antigen complexes typically show contact interfaces of 6-9 nm2, involving 14-21 residues on each side [90-92]), would on average overlap one such ligand that is resident in a bacterial surface pressed against it. If there are 100 such arrays uniformly distributed over the entire nanorobot surface, then a randomly chosen mutual contact area of only 1% of the nanorobot surface suffices to ensure that there is at least one array overlapping a unique ligand on the bacterial surface during a collision. Of course, the probability of binding, even given mutual contact, is not unity, but perhaps only ~10% (e.g., Nencounter ~ 10 [1, LINK]). However, this factor is almost completely offset because there are nine equivalent array sets -- one set for each of the nine unique bacterial ligands -- and recognition and binding of any one of the nine unique ligands will suffice to bind the bacterium securely to the nanorobot. Since array members need not be adjacent, the actual physical configuration on the microbivore surface is a bit different. The binding sites are modeled after the narrowband chemical sensor described in Nanomedicine [1, LINK], Figure 4.2. Each 3×3 receptor block consists of nine 7 nm × 7 nm receptor sites, one for each of the nine species-specific bacterial coat ligands. There are 20,000 of these 3×3 receptor blocks distributed uniformly across the microbivore surface. Each 3×3 receptor block measures 21 nm × 21 nm ×10 nm. A single receptor, if bound to a ligand, may provide a binding force of 40-160 pN [1, LINK], probably larger than the largest plausible in sanguo dislodgement force of ~100 pN [1, LINK] and thus gripping the bacterium reasonably securely. The recognition event can be consumated in tmeas ~ 30 microsec, according to Eqn. 8.5 from Nanomedicine [1, LINK]. As an operational procedure, once any one of the nine key ligands has been detected, all of the remaining unoccupied receptors for that ligand in other receptor blocks can be deactivated, and so on until all nine ligands have been individually confirmed -- a combination lock whose completion triggers bacteriocide. Interestingly, during phagocytosis by macrophages most injected particles are recognized by more than one receptor; these receptors are capable of cross-talk and synergy, and phagocytic receptors can both activate and inhibit each other's function [247]. Microbial binding is energetically favored; if
binding energy is ~240 zJ per microbial ligand [1,
LINK] (1
zeptojoule (zJ) = 3.1.2 Telescoping GrapplesOnce the target bacterium has been confirmed and temporarily secured to the microbivore surface at >9 points with a minimum binding force of >360-1440 pN, telescoping robotic grapples emerge from silos in the nanodevice surface to establish secure anchorage to the microbe's plasma membrane or outer coat. Each grapple is mechanically equivalent to the telescoping robotic manipulator arm described by Drexler [93], but 2.5 times the length. This manipulator when fully extended is a cylinder 30 nm in diameter and 250 nm in length with a 150-nm diameter work envelope (to the microbivore hull surface), capable of motion up to 1 cm/sec at the tip at a mechanical power cost of ~0.6 pW at moderate load (or ~0.006 pW at 1 mm/sec tip speed), and capable of applying ~1000 pN forces with an elastic deflection of only ~0.1 nm at the tip. (Interestingly, supplementing chemispecificity (Section 3.1.1) gram-negative bacteria can be distinguished from gram-positive organisms by their wavy surface appearance when scanned by AFM [94], a subtle morphological difference that should also be detectable by grapple-based pressure sensors that could help confirm microbial identity.)Each telescoping grapple is housed beneath a self-cleaning irising cover mechanism that hides a vertical silo measuring 50 nm in diameter and 300 nm in depth, sufficient to accommodate elevator mechanisms needed to raise the grapple to full extension or to lower it into its fully stowed position. At a 1 mm/sec elevator velocity, the transition requires 0.25 millisec at a Stokes drag power cost (operating in human blood plasma) of 0.0008 pW, or 0.008 pW for 10 grapples maximally extended simultaneously [1, LINK]. The elevator mechanism consists of compressed nitrogen gas rotored into or out of the subgrapple chamber volume from a small high-pressure sealed reservoir, a pneumatic piston providing the requisite extension or retraction force. A grapple-distension force of ~100 pN applied for a distance of 250 nm could be provided by 25 atm gas pressure in a minimum subgrapple chamber volume of 104 nm3, involving the importation of ~6000 gas molecules. Removal of these ~6000 gas molecules from a maximum subgrapple chamber volume of 105 nm3 provides a ~1 atm pressure differential and a maximum grapple-retraction force of ~100 pN; cables or other mechanisms may assist in retraction if more force is needed. The aperture of the irising silo cover can be controlled to continuously match the width of the protruding grapple, greatly reducing the intrusion of foreign biomolecules into the silo. Each grapple is terminated with a reversible footpad ~20 nm in diameter. In the case of gram-positive bacteria, a footpad may consist of 100 close-packed lipophilic binding sites targeted to plasma membrane surface lipid molecules, providing a secure 1000 pN anchorage between the nanorobot and the bacterium assuming a single-lipid extraction force of ~10 pN [1, LINK]. In the case of gram-negative bacteria, a footpad with binding sites for ~3 murein-linked covalently attached transmembrane protein molecules would provide a secure 120-480 pN anchorage, assuming 40-160 pN/molecule and ~9 such molecules per 1000 nm2 of microbial surface (Section 3.1.1). In either case, undesired adhesions with bacterial slime must be avoided. The footpad tool is rotated into, or out of, an exposed position from behind a protective cowling, using countercoiled internal pull cables. The tiniest bacterium to be digested may be ~200 nm in diameter (Section 2.1.4), but the smallest virus can be only ~16 nm wide (Section 2.2). Since the work envelopes of adjacent grapples picking particles bound to the hull surface extend 150 nm toward each other from either side, the maximum center-to-center intergrapple separation that permits the ciliary transport of 16 nm objects is ~300 nm. This requires 1 grapple per 0.09 micron2 of nanorobot surface, for a total of 277 grapple silos uniformly distributed over the entire 26.885 micron2 microbivore outer hull, excluding the two 1-micron2 port doors. (One or more grapple-containing bridges across the annular exhaust port aperture (Section 3.1.4) may be necessary if it is desired to transport targets <200 nm in diameter from the circular DC exhaust port island to the main grapple field of the microbivore, allowing subsequent transport to the ingestion port inlet; such bridges are not included in the present design.) During transport, a bacterium of more typical size such as a 0.4 micron × 2 micron P. aeruginosa bacillus may be supported by up to 9 grapples simultaneously. A somewhat larger E. coli bacterium would be supported by up to 12 grapples. After telescoping grapples are securely anchored to the captive bacterium, the receptor blocks are debonded from the microbial surface, leaving the grapples free to maneuver the pathogen as required. Grapple force sensors inform the onboard computer of the captive microbe's footprint size and orientation. The grapples then execute a ciliary transport protocol in which adjacent manipulators move forward and backward countercyclically, alternately binding and releasing the bacterium, with new grapples along the path ahead emerging from their silos as necessary and unused grapples in the path behind being stowed. Manipulator arrays, ciliary arrays (MEMS), and Intelligent Motion Surfaces are related precursor (and currently available) technologies (reviewed in Section 9.3.4 of Nanomedicine [1, LINK]). Rodlike organisms are first repositioned to align
their major axis perpendicular to a great circle plane containing
both the device center point and the ingestion port at the front of
the device. This keeps the organism traveling over surfaces having
the largest possible radius of curvature during transport, thus
minimizing any forces necessary to bend the bacterium as it follows
the curved microbivore surface. A cylindrical bacterium of length
Ltube and bending stiffness
ktube is bent by a force
F into a circle segment having
radius of curvature Rcurve ~
(ktubeLtube2
/ 2 F) for small deflections.
For the bacillus P. aeruginosa, Ltube
~ 2 microns and tube radius is ~0.2 microns; the elastic modulus is
2.5 ×107 N/m2 for the 3-nm thick hydrated
sacculus [97],
giving ktube Organisms of all shapes are conveyed toward the ingestion port via cyclical ciliary cycling motions. At a transport velocity of 1 mm/sec, a microbe captured at the greatest possible distance from the ingestion port (~3 microns) is moved to the vicinity of the ingestion port in ~3 millisec. The Stokes law energy cost of transporting an E. coli bacterium through blood plasma side-on at 1 mm/sec is 0.01 pW, so transport power is dominated by mechanical losses in the grapples, a total of ~0.06 pW if 10 grapples are operated simultaneously. Because the ingestion port is slightly recessed
into the body of the nanorobot ellipsoid at the equator, the
approaching bacterium must be carried around an inlet rim having a
considerably smaller radius of curvature than the main body of the
microbivore. The inlet rim is essential in this design and provides
needed mechanical control from inlet-wall grapples as the microbe is
fed into the ingestion port. From simple geometry, if one grapple is
fully extended to length L =
Lgrap and the adjacent
grapple is almost fully retracted to length
L ~ 0, then the bacillus can be
conveyed around an inlet rim curve of radius Rrim
with zero bending if the distance between the adjacent grapples is
no more than dmax ~ 2
Rrim 3.1.3 Ingestion Port and Morcellation ChamberThe ingestion port door is an oval-shaped irising mechanism [1, LINK] with an elliptical aperture measuring 0.8654 microns × 1.4712 microns, providing a 1 micron2 aperture when fully open. Assuming 0.5 micron2 of contact surfaces sliding ~1 micron at 1 cm/sec, power dissipation is ~3 pW during the 0.1 millisec door opening or closing time. To allow handing small particles like viruses securely into the ingestion port, the porthole mechanism can be programmed to iris open in an off-center manner if required. For example, if manipulating a small virion particle the hole's center should initiate within 150 nm of a sidemost edge of the port (i.e., within one grapple surface-reach distance, either left or right side); after the growing aperture reaches the edge of the nearest side, it can then continue to dilate toward the edge on the opposite side while retaining its expanding elliptical shape. On the other hand, if a bacterium >~0.632 microns in diameter is being manipulated, the port door may be programmed to iris open from the center. During internalization the port doors perform gentle test-closings, with associated force sensors providing feedback as to the completeness of the internalization process and enabling the microbivore to detect the pinch points of linked bacilli to allow separation at these points, if necessary. In the case of motile bacilli having long flagellar tails, the premature closing of the ingestion port door may sever the tail, casting the immunogenic tail fragment adrift in the blood; this outcome must be avoided (Section 4.3).Opening the ingestion port door allows entry into the morcellation chamber (MC), a cylindrical chamber 2 microns in length and the same interior elliptical cross-section as the port door, giving a total open volume of 2 micron3 which is large enough to hold one intact microorganism because most sepsis-related bacteria are <2 micron3 in volume (Table 1). Recessed into the MC walls are 10 diamondoid cutting blades (possibly multisegmented), each ~2 micron long, ~0.25 micron wide, and 10 nm thick with a 1 nm cutting edge, giving ~0.050 micron3 of blades (~0.005 micron3/blade). Following the analysis of nano-morcellation systems described elsewhere [1, LINK], to mince material having Young's modulus ~108 N/m2 using one blade at a time (reserving the other 9 blades as replacements or to provide alternative chopping geometries) requires the application of ~100 nN/chop, consuming up to ~100 pW during a process in which the blade reciprocates at 50 Hz and travels at ~60 micron/sec, making 20 cuts in a total mincing time of 400 millisec. (Bacterial walls include a 3-6 nm thick hydrated sacculus [97] and include a cross-linked peptidoglycan (murein) mesh [95-97] with strands spaced ~1.3 nm apart [98].) The resulting morcellate should consist largely of organic chunks ~3-10 nm in diameter [1, LINK]. An intriguing alternative configuration is a diamondoid sieve or dragnet that could be pulled repeatedly through the MC, analogous to pushing the microbe forcibly through a strainer; other possible fragmentation techniques such as sonication appear to require too much onboard acoustic energy to be feasible (e.g., power intensities of ~106 pW/micron2 [1, LINK]). Although complex mechanical assemblages may dissipate 109 W/m3, mechanomechanical and electromechanical transducers are generally very efficient, dissipating 1012-1016 W/m3 during mechanical energy transmission [1, LINK; 93]. Conservatively assuming that the nanomotors needed to drive the chopping blade may dissipate ~1010 W/m3, then a ~0.01 micron3 drive motor is required to operate the blade; we allocate a total of 0.1 micron3 for multiple drive motors, thus providing tenfold redundancy. Another 0.1 micron3 is allocated for blade housings. A diamondoid MC wall ~10 nm thick (materials volume ~0.073 micron3) allows the MC to withstand internal pressures >1000 atm, far higher than the natural internal microbial pressurization of 3-5 atm [99]. (Bacterial rigidity is regulated by turgor pressure [100].) Once microbial mincing is complete, the morcellate must be removed to the digestion chamber (Section 3.1.4) using an ejection piston. A 20-nm thick piston pusher plate driven by a 2 micron long, 10 nm thick pusher cable (energized by the chopping blade motor coupled through a mechanical transmission gearbox) comprises ~0.02 micron3 of device volume. This piston moves forward at ~20 microns/sec, applying ~1 atm of pressure to push morcellate of viscosity ~100 kg/m-sec through a 1 micron2 gated annular aperture for a chamber length of 2 microns, emptying the MC in ~100 millisec with a Poiseuille fluid flow power dissipation [1, LINK] of ~2 pW. Interestingly, the energy dissipation rate required to disrupt the plasma membrane of ~95% of all animal cells transported in forced turbulent capillary flows is on the order of 108-109 W/m3 [101], corresponding to a mechanical power input of 100-1000 pW into a 1 micron3 chamber volume. The annular MC/DC interchamber door must be opened before activating the MC ejection piston; its size and power specifications are similar to those of the annular DC exhaust port door (Section 3.1.4.4). The MC ejection piston also is used initially to draw the microbe into the MC in a controlled manner. By slowly pulling a vacuum after the ingestion port door has opened, the piston can apply ~1 atm of negative pressure over the ~1 micron2 leading surface of the bacterium, or up to ~100 nN of force. The Poiseuille flow of a microorganism of viscosity ~1000 kg/m-sec through a 1 micron2 aperture with a 1 atm pressure differential into a chamber 2 microns in length dissipates 0.2 pW as the bacterium is drawn into the chamber at a speed of 2 microns/sec, thus requiring ~1 second for complete internalization of 2 micron3 of ingesta. 3.1.4 Digestion Chamber and Exhaust PortThe digestion chamber (DC), like the MC, has a total open volume of 2 micron3. The DC is a cylinder of oval cross-section surrounding the MC, measuring roughly 2.0 microns in width, 1.3 microns in height, and 2.0 microns in length, with a mean ~0.5 micron clearance between the DC and MC walls and a materials volume of 0.11 micron3 assuming diamondoid walls ~10 nm thick. Morcellate is pumped from the MC into the DC where a preprogrammed sequence of engineered enzymes are successively injected and extracted, reducing the morcellate primarily to monoresidue amino acids, mononucleotides, free fatty acids and monosaccharides, which are then harmlessly discharged into the environment.If the morcellate consists of organic chunks ~3-10 nm in diameter (Section 3.1.3), enzymes directed against specific bond types may attack these bonds only if they are exposed on the outermost surface of each chunk. Considering for simplicity only proteinaceous chunks, and given that the average amino acid has a molecular weight of 141.1 daltons and a molecular volume of Vres ~ 0.49 nm3, then a chunk of volume Vchunk may be regarded as having Nlayer successive surface layers where Vchunk ~ Vres (1 + 2Nlayer)3. Taking Vchunk1/3 = 10.2 nm for the largest pieces implies a chunk comprised of 2197 residues and having Nlayer ~ 6 layers that must be processed sequentially, like peeling an onion one skin at a time. Thus the entire enzyme suite must be shuttled in and out of the DC six times, with one "layer" of all chunks being processed during each of the six subcycles. 3.1.4.1 Artificial Enzyme SuiteArtificial digestive enzymes may be designed to attack just one class of chemical bond [102]. For example, the natural serine protease enzyme chymotrypsin only cleaves peptide bonds at the carboxylic ends of residues having large hydrophobic side chains, such as the aromatic amino acids phenylalanine, tryptophan, and tyrosine [103, 104]. The proteolytic enzyme trypsin exhibits a different specificity, cleaving peptide bonds on the C-terminal side of the basic residues arginine and lysine [103]. The endopeptidase elastase attacks bonds adjacent to small amino acid residues such as alanine, glycine, and serine [105] and will cleave tri-, tetra-, and penta-peptides of alanine [104]. Enzymes which will cleave the unusual right-handed (D-enantiomeric) amino acids found in bacterial coats, including D-aminopeptidase [106] or D-stereospecific amino-acid amidase [107], D-peptidase and DD-peptidase [107], carboxypeptidase DD [108] and D-amino acid acylase [109] are well-known.To prevent self-digestion during storage and use, each artificial peptidase is engineered so that the class of residue it is designed to attack is not exposed on its own external physical surface [112] -- that is, each artificial enzyme minimally exhibits strong autolysis resistance [110-116], with an ideal objective of near-zero autolysis. (A few natural enzymes retain full post-autolysis functionality [117].) Another significant design constraint is that natural bacterial enzymes already present in the morcellate (e.g., elastase produced by P. aeruginosa [118]) must have negligible activity against any of the microbivore's artificial enzymes. Since the target microbe's enzyme inventory is known in advance, the microbivore enzyme suite can be tailored to deal with any unusually troublesome bacterial enzymes, and optimal pH in the DC can be actively managed (see below). Ensuring biological digestive universality while allowing the enzyme engineer sufficient diversity of available protein building blocks requires a minimum of two pre-activated artificial enzymes that attack specific peptide bonds in each of the seven major amino acid classes -- acidic (Asn, Asp, Gln, Glu), aliphatic (Ala, Gly, Ile, Leu, Val), aromatic/hydrophobic (His, Phe, Trp, Tyr), basic (Arg, His, Lys), hydroxylic (Ser, Thr, Tyr), imino (Pro), and sulfur (Cys, Met). The present design thus includes a requirement for 14 artificial endopeptidases, plus 2 broad-spectrum artificial tripeptidase [119] and dipeptidase [120] if needed to complete the digestion of potentially bioactive tripeptides and dipeptides to free amino acids. Enzymes capable of degrading nucleic acid polymers are classified as deoxyribonucleases (specificity for DNA) or ribonucleases (specifically hydrolyzing RNA), or as exonucleases (hydrolyzing a nucleotide only when present at a strand terminus, moving in only one direction, either 3'®5' or 5'®3') or endonucleases (cleaving internal phosphodiester bonds to produce either 3'-hydroxyl and 5'-phosphoryl termini or 5'-hydroxyl and 3'-phosphoryl termini) [105]. Some endonucleases can hydrolyze both strands of a double-stranded molecule, others attack only one strand of a double-stranded molecule, while still others cleave only single-stranded molecules. Restriction endonucleases recognize specific DNA sequences -- for example, Hpa I recognizes a specific double-strand 6-base sequence (GTTAAC/CAATTG) and selectively cleaves both strands of the double strand in the middle at the TA/AT bond, producing an unreactive molecular "blunt end" [105]. There are ten distinct dinucleotide bond combinations (AA, AC, AG, AT, CC, CG, CT, GG, GT, and TT), which suggests that 10 artificial endonucleases may suffice, plus 2 general-purpose dinucleases to complete the digestion to mononucleotides, for a total of 12 artificial polynucleotidases. Additional engineered enzymes (not included in the present design) may be needed to digest bacteriophages that may be resident inside certain bacteria. To avoid digestion by bacterial restriction enzymes, phages often employ unusual molecular substitutions involving 2,6-diaminopurine, 6-methyladenine, 8-azaguanine, 5-hydroxymethyl uracil, 5-methylcytosine, 5-hydroxymethylcytosine, and others [121]. For example, B. subtilis phage DNA replaces thymine with hydroxymethyluracil and uracil; S-2L cyanophage replaces adenine by 2-aminoadenine (2,6-diaminopurine); SPO1, SP82G, and Phi-e substitute hydroxymethyl dUTP for dTTP in the phage DNA up to 20%; PBS1 and PBS2 phages substitute uracil for thymine; T-even (T2/T4/T6) phage DNA replaces dCMP by hydroxymethylcytosine which is then further glycosylated, rendering the phage DNA resistant to host restriction; and in phage Mu DNA, a unique glycinamide moiety modifies about 15% of the adenine residues [121]. Given our complete future knowledge of phage genomes and the bacteria they are likely to inhabit, a comprehensive phage digestive strategy can be planned and installed in advance, during microbivore design and construction. This problem is not considered serious in the case of standard antibiotic therapy. Free adenosine (a mononucleotide) is involved in the regulation of coronary blood flow [122], and certain free nucleotides have been shown to exhibit minor physiological action on lymphocytes [123] and T cells [124] in animal models, so additional nucleotidases, phosphatidases and nucleosidases may be added if necessary to reduce free mononucleotides to phosphoric acid, sugars, and purine/pyrimidine bases prior to discharge from the nanorobot. However, such additional enzymes are not included in the present microbivore design because nucleotidase is naturally present in normal human serum [125-129] and at elevated serum levels in many disease conditions [129-133]. Microbial lipids may be digested by analogs of pancreatic lipase (e.g., steapsin) or lipoprotein lipase which hydrolyze polyacylglycerols (mostly glycosyl diacylglycerols in bacteria) containing fatty acid chains into free fatty acids and glycerol, by cholesterol esterase that hydrolyzes cholesteryl esters into free cholesterol (although cholesterol and other sterols are relatively rare in microorganisms [134-136]), by phospholipase that attacks phospholipids producing glycerol, fatty acids, phosphoric acid, and perhaps choline [105], or by sphingolipidases [137] or ceramidases [138] that hydrolyze the sphingolipids found in some bacteria, resulting in mostly glycerol and saturated (in bacteria) free fatty acids in the final digesta. Acyloxyacyl hydrolase removes the secondary (acyloxyacyl-linked) fatty acyl chains from the lipid A region of bacterial lipopolysaccharides (LPS endotoxin), thereby detoxifying the molecules [139]. The present microbivore design assumes a requirement for 5 artificial lipases. Microbial carbohydrates may be digested by an amylase that hydrolyzes starch and glycogen, and by a selection of oligosaccharidases (e.g., maltase, sucrase-isomaltase) and disaccharidases or saccharases (e.g., lactase, invertase, sucrase, trehalase) to complete the digestion to monosaccharides [105]. (Lactase also has a second active site for splitting glycosylceramides [105].) The present design assumes a requirement for 4 artificial carbohydrases in the microbivore enzyme suite. Finally, simple anions or cations may be required for pH management of the morcellate, and 25% of all enzymes contain tightly bound metal ions or require them for activity [105], most commonly Mg++, Mn++, Ca++, or K+; certain low-bioavailability but essential cofactors such as iron and copper might also need to be actively managed. It might also be necessary in some cases to inject and extract small quantities of superoxide dismutase, catalase and chelating agents such as metallothionein, ferritin, or transferrin to control potentially damaging concentrations of superoxides and metals in the morcellate, or small quantities of other specialized enzymes analogous to heme oxygenase, biliverdin reductase and beta-glucuronidases to digest bacterial porphyrins [244], enzymes [245] to cleave bacterial rhodopsins, and so forth, but a full analysis of these factors is beyond the scope of this paper. The present design assumes a requirement for 3 additional chemical species of this type, to be manipulated simultaneously with the artificial enzymes as previously described. Full digestion of the morcellate, constituting one complete digestion cycle, is thus presumed to require six subcycles of activity, with each subcycle involving the serial injection and extraction of 40 different enzymes or enzyme-related molecules (i.e., 40 sub-subcycles per subcycle), one after the other, for a total of 240 enzyme sub-subcycles. Interestingly, intracellular lysosomes are known to contain ~40 digestive enzymes capable of degrading all major classes of biological macromolecules -- including at least 5 phosphatases, 4 proteases, 2 nucleases, 6 lipases, 12 glycosidases, and an arylsulfatase [140, 141]. 3.1.4.2 Digestion Cycle TimeThe duration of each enzyme sub-subcycle depends primarily upon two factors: (1) the speed of enzymatic action (Section 3.1.4.2.1), which may differ somewhat for each enzyme and each substrate, and (2) the speed at which enzymatic molecules can be rotored into and out of the DC (Section 3.1.4.2.2).3.1.4.2.1 Speed of Enzymatic ActionIf enzyme molecules are plentiful and substrate molecules are rare (typically 1%-100% of the enzymes), the most appropriate measure of enzymatic speed is the enzymatic efficiency (kcat / Km) = 1.5-28 ×107 molecules of substrate converted to product per second, per molar concentration of enzyme, for a wide variety of enzymes [142]. Here, the Michaelis constant Km is the substrate concentration that produces the half-maximal reaction rate, and kcat is the reaction rate in product molecules generated per unit time per enzyme molecule.However, for most of the digestion cycle the DC
environment consists of a relatively small number of temporarily
resident enzyme molecules floating in a sea of plentiful substrate.
Zubay [142]
notes that in this situation, the speed of enzymatic action is
considerably slower and kcat,
also known as the enzyme turnover number, is the most relevant
measure of enzyme catalytic activity.
Table
3 shows that for peptidases, kcat
ranges from
To estimate the time required for each enzymatic sub-subcycle, for simplicity the initial morcellate of volume Vmorc ~ 2 micron3 is assumed to consist mostly of water containing a volume fraction fprot ~ 0.30 (30%) of now-minced protein. The specific volume of the average amino acid residue is taken as Vres ~ 0.49 nm3/residue and the required number of enzymatic sub-subcycles is taken as Nessc ~ 240. Then the average number of peptide bond scissions per sub-subcycle is Nbondx = (Vmorc fprot) / (VresNessc) ~ 5 ×106 bonds/sub-subcycle, and the processing time per sub-subcycle is tenz ~ Nbondx / (kcatnenz) where nenz is the number of enzyme molecules injected into the morcellate during each sub-subcycle. Taking nenz = 104 enzyme molecules and kcat = 104 Note that the diffusion time required by an enzyme
molecule of radius 3.47 nm at 37°C in a plasma-like fluid of
viscosity 3.1.4.2.2 Speed of Enzyme-Transport RotorsIf nenz enzyme molecules must be transferred during each sub-subcycle in a transport time ttransport using nrotor molecular sorting rotors with each rotor operating at a constant transport rate of krotor molecules/rotor-sec, then nrotor = nenz / (ttransportkrotor). Each artificial enzyme molecule is assumed to consist of ~350 residues with a molecular weight of ~50 kDa and a molecular volume of ~175 nm3, giving a molecular diameter of ~6.9 nm if assumed spherical. Taking the excluded volume per enzyme molecule binding site as 7 nm in diameter, a sorting rotor 8 nm thick with 10 receptors plus one 8-nm blank space per rotor requires an enzyme-transport rotor circumference of 78 nm, giving a rotor diameter of 25 nm and a rectangular face area and volume per rotor of ~200 nm2 and ~5000 nm3, respectively [1, LINK; 93].What is the value of krotor
during enzyme extraction? The injection of 104 enzyme
molecules into the 2 micron3 digestion chamber produces
an enzyme concentration of However, increasing nrotor
to 2000 rotors to provide tenfold redundancy, while holding
textract constant, reduces
the required krotor by a
factor of 10 -- e.g., to kr(10,000)
~ 0.1 molecule/rotor-sec. According to
Section 3.2.2
of Nanomedicine [1,
LINK],
the diffusion current to a rotor of face area 200 nm2
(equivalent circular radius ~8 nm), taking the enzyme diffusion
coefficient as Increasing nrotor to 2000 rotors per enzyme species also permits the elimination of enzyme storage tanks and associated support structures, because 2 ×104 enzyme molecules can be stored in 2000 rotors each having 10 enzyme receptor sites per rotor. If the rotors are turned at 1 kHz, the entire enzyme inventory is injected into the DC in ~1 rotor rotation time, giving tinject ~ 1 millisec. 3.1.4.3 Summary of Digestion SystemsDuring each sub-subcycle, 104 enzyme molecules are injected into the digestion chamber in tinject ~ 1 millisec (Section 3.1.4.2.2). Enzymatic digestive action then commences, requiring tenz ~ 50 millisec to go to completion (Section 3.1.4.2.1). The 104 enzyme molecules are then extracted from the DC and returned to the in-rotor reservoir in textract ~ 50 millisec (Section 3.1.4.2.2). Total processing time per sub-subcycle is tssc ~ 101 millisec, so one complete microbivore digestion cycle comprising 240 sub-subcycles requires ~24.24 sec.There is one set of 2000 enzyme-transport rotors for each of the 40 enzyme species transported, hence there are 80,000 enzyme-transport rotors protruding into the DC. These rotors have a total face area of 16 micron2, somewhat more than the ~10 micron2 cylindrical DC sidewall area, thus require some slight rotor invagination into the DC volume. The rotors occupy a total onboard volume of 0.4 micron3 with an additional 0.1 micron3 allocated for drive mechanisms, housings, and other rotor-related support, for a total 0.5 micron3 enzyme-transport rotor volume allocation. If the binding energy of each enzyme receptor is ~240 zJ [1, LINK], then the total energy cost to eject 104 enzyme molecules from their rotors is ~0.0024 pJ, representing a mean power requirement of 2.4 pW when injection is performed over tinject ~ 1 millisec. Rotor drag power during extraction is negligible, so full-cycle power consumption averages ~0.024 pW. Note that bond hydrolysis is often thermodynamically favored, evolving a free energy of hydrolysis Ehydrol ~ -4 zJ/bond to -14 zJ/bond for breaking peptide bonds [164, 165], -21 zJ/bond to -46 zJ/bond for glycosides and sugars [165], and -15 zJ/bond to -103 zJ/bond for various organophosphate bonds [165, 166]. Hence the scission of Nbondx ~ 5 ×106 bonds/sub-subcycle during a time tssc ~ 101 millisec/sub-subcycle produces a continuous digestive waste heat of Pdigest = EhydrolNbondx / tssc~ 0.2-5 pW per nanorobot, but most likely <1 pW for typical microbial compositions. It is well-known that protein components of the cell membrane are continually removed and replaced, with the turnover rate in the unprotected cellular environment varying for different proteins but averaging a half-life of ~200,000 sec or ~ 2 days [140, 141]. However, each enzyme spends a total time of 0.306 sec per digestion cycle (Table 6) exposed to the morcellate or intermediate digesta, which suggests useful enzyme suite lifetimes of at least 104-105 digestion cycles (e.g., mission lifetimes >3-30 days assuming continuous digestive activity) conservatively may be expected. In typical clinical deployments to combat acute bacteremia, each microbivore will experience at most 1-10 digestion cycles during the entire mission. Additionally, artificial enzymes that are deployed in relatively nondegradative controlled intrananorobotic environments might be expected to survive perhaps an order of magnitude longer than natural enzymes in the wild. This increased survivability, coupled with the tenfold redundancy of all critical onboard systems including the artificial enzymes and their transport mechanisms, suggests that extended microbivore missions lasting many months in duration might be feasible. 3.1.4.4 Ejection Piston and Exhaust PortOnce microbial digestion is complete, the digesta must be discharged into the external environment of the nanorobot. Egestion is achieved using an annular-shaped ejection piston comprised of a 20-nm thick piston pusher plate driven by at least two 2-micron long, 10-nm thick pusher cables, comprising ~0.02 micron3 of device volume. This piston moves forward at ~200 micron/sec, applying ~0.1 atm of pressure to push digesta of viscosity <1 kg/m-sec through a 1 micron2 gated annular exhaust port, through a distance of the 2-micron DC length, emptying the DC in ~10 millisec with a Poiseuille fluid flow power dissipation [1, LINK] of ~2 pW. Afterwards, the piston is retracted, effectively pulling a vacuum in the DC in preparation to receive the next batch of morcellate from the MC.An annular exhaust port door must be opened prior to activation of the ejection piston to allow the digesta to escape. The exhaust port door is an oval-shaped irising mechanism [1, LINK] with an annular elliptical aperture measuring 0.721 microns × 1.227 microns along the inside curve and 1.108 microns × 1.884 microns along the outside curve in vertical plane projection, providing a 1.161 micron2 aperture in the hull surface when fully open. Assuming 0.5 micron2 of contact surfaces sliding ~1 micron at 1 cm/sec, power dissipation is ~3 pW during the 0.1 millisec door opening or closing time. 3.2 Microbivore Support SystemsVarious mechanical subsystems are required to support the principal activities of the microbivore digestive system. These support subsystems include the power supply (Section 3.2.1), external and internal sensors (Section 3.2.2), the onboard computer (Section 3.2.3), structural support (Section 3.2.4), and a ballast system to permit nanapheresis (Section 3.2.5).3.2.1 Power Supply and Fuel Buffer TankageThe microbivore is scaled for a maximum power output of 200 pW. The power source is assumed to be an efficient oxyglucose powerplant such as a fuel cell, with net output power density of ~109 W/m3 [1, LINK]. Each powerplant thus requires an onboard volume of 0.2 micron3. Ten powerplants (each one independently capable of powering the entire nanorobot at its maximum power requirement) are included onboard for redundancy, giving a total powerplant volume requirement of 2 micron3.The microbivore is initially charged with glucose and compressed oxygen (stored in sapphire-walled tankage), and thereafter absorbs its ongoing requirements directly from the bloodstream. Assuming 50% energy conversion efficiency and a 200 pW continuous power production requirement, each glucose and oxygen molecule that are consumed produce 2382.5 zJ or 397.1 zJ, respectively [1, LINK], indicating a peak burn rate of 8.4 ×107 molecules/sec of glucose and 50 ×107 molecules/sec of O2. The minimum glucose concentration in normal adult
human blood is The minimum free molecular oxygen concentration in
normal adult human blood is Waste products from oxyglucose power generation include water and carbon dioxide. There are 50 ×107 molecules/sec of each waste species produced, which may be ejected from the nanorobot using 500 standard sorting rotors for each species, assuming a transport rate of ~106 molecules/rotor-sec. The present design thus employs 500 rotors each for H2O and for CO2, for each of the ten independent powerplants. However, in an emergency these wastes could alternatively be bulk-vented to the external environment without harmful effect -- the effervescence limit for point releases of bulk CO2 in arterial plasma is ~70 ×107 molecules/sec [1, LINK]. The microbivore design thus includes 86,000 small-molecule sorting rotors for energy-molecule transport with full tenfold redundancy, occupying a total of ~8.6 micron2 of microbivore surface area and 0.103 micron3 of microbivore volume. Energy dissipation by the rotor system, if operated at the maximum 200 pW production rate, is 16 pW assuming the transfer of 158.4 ×107 molecules/sec at an energy cost of ~10 zJ/molecule (net energy cost after compression energy recovery) [1, LINK]. On the microbivore surface, the energy-molecule transport rotors are arranged as compactly as possible into ten lune-shaped sectors (one for each of the ten powerplants) running from front to back (i.e., from ingestion port to exhaust port), with 8600 rotors/lune. Onboard oxyglucose fuel tanks are scaled to provide a buffer supply of ~one-half circulation time or one digestion cycle time (~30 sec) of peak device energy requirement. Assuming a 50% aqueous solution of glucose in the glucose storage tank and a molecular volume of 0.191 nm3/molecule for glucose molecules [1, LINK], then the required glucose tank volume is 0.962 micron3 to hold a buffer supply of 252 ×107 molecules of glucose fuel. Adding ~0.038 micron3 for 5-nm thick diamondoid walls and other support structure gives a 1.0 micron3 microbivore volume requirement for the glucose buffer tank. Assuming oxygen storage at 1000 atm (0.0791 nm3/molecule [1, LINK]), the 30-sec buffer supply of 1500 ×107 oxygen molecules at 200 pW peak powerplant output requires an oxygen tank of volume 1.187 micron3. A spherical pressure tank requires a diamondoid wall thickness of >3.3 nm to avoid bursting; the present design assumes 10 nm thick tank walls. Adding ~0.055 micron3 for tank material volume and 0.058 micron3 for other support structure gives a 1.3 micron3 microbivore volume requirement for the oxygen buffer tank. Diamondoid mechanical cables may transmit internal
mechanical energy at power densities of ~6 ×1012 W/m3
[1,
LINK].
Therefore a single cable that can transmit the entire microbivore
power output of 200 pW may have a volume of 3.2.2 SensorsThe microbivore needs a variety of external and internal sensors to complete its tasks. External sensors include chemical sensors for glucose, oxygen, carbon dioxide, and so forth, up to 10 different molecular species with 100 sensors per molecular species. Each 10 nm × 45 nm × 45 nm chemical concentration sensor with 450 nm2 face area is assumed to discriminate concentration differentials of ~10% and displace ~105 nm3 of internal nanorobot volume [1, LINK]. Taking chemical sensor energy cost as ~10 zJ/count [1, LINK] with ~104 counts/reading [1, LINK], then 10 readings/sec by each of 1000 microbivore sensors gives a maximum sensor power requirement of ~1 pW by a chemical sensor facility that displaces a total of ~0.1 micron3 of device volume and 0.45 micron2 of device surface area.Acoustic communication sensors mounted within the
nanorobot hull permit the microbivore to receive external
instructions from the attending physician during the course of in
vivo activities. Assuming (21 nm)3 pressure transducers [2,
LINK], then 1000 of these transducers displace ~0.01 micron3
of device volume and 0.44 micron2 of device surface area,
producing a small net power input to the device of An internal temperature sensor capable of
detecting 0.3°C temperature change [1,
LINK] may
have a volume of (~46 nm)3 ~ 3.2.3 Onboard ComputersStarting with Drexler's benchmark (400 nm)3 gigaflop mechanical nanocomputer [93], the microbivore computer is scaled as a 0.01 micron3 device in principle capable of >100 megaflops but normally operated at <~1 megaflop to hold power consumption to <~60 pW. Assuming ~5 bits/nm3 for nanomechanical data storage systems [93] and a read/write cost of ~10 zJ/bit at a read/write speed of ~109 bits/sec [1, LINK; 93], then 5 megabits of mass memory to hold the microbivore control system (Table 4) displaces a volume of 0.001 micron3 and draws ~10 pW while in continuous operation. The current microbivore design includes ten duplicate computer/memory systems for redundancy (with only one of the ten computer/memory systems in active operation at a time), displacing a total of 0.11 micron3 and consuming <~70 pW.
3.2.4 Structural SupportThe external microbivore hull is taken as a 50-nm thick diamondoid surface of surface area 24.885 micron2 (again excluding the 2 micron2 of ports), a materials volume of 1.2443 micron3. The buckling pressure of a circular diamondoid cylinder of similar dimensions, subjected to crushing forces, is ~300 atm. However, an ellipsoidal hull is considerably weaker than a circular hull so some internal cross-bracing (not included in the present design) might be necessary to resist the ~50 atm force of dental grinding [1, LINK; 2, LINK].An additional 0.3799 micron3 of unspecified mechanisms and support structure are included in the present design, which is summarized in Table 5.
3.2.5 Ballasting for NanapheresisAs in previous designs [2, LINK], the microbivore can alter its overall density to achieve approximately neutral buoyancy, thus allowing convenient removal from the patient's body via nanapheresis [1, LINK] after the therapeutic purpose is complete. (More elegant methods for nanorobot ingress and egress from the human body are readily imagined but are beyond the scope of this scaling design study.) Density is altered by exhausting the onboard O2 buffer tank and then pistoning the MC and DC empty, thus establishing a vacuum in both chambers. If either or both of the pistons have failed, the device can still be prepared for nanapheresis by venting the compressed oxygen into the MC and DC, blowing the two chambers clear of fluid and filling this volume with gas, which is substantially similar in density to vacuum from the standpoint of ballasting.Assuming a mean density of 1900 kg/m3
for diamondoid nanomechanical structure, the "dry weight" of a
microbivore is ~12.2 pg, giving a minimum achievable density of
~1000 kg/m3. The density of a fully charged microbivore
with both chambers loaded is ~17.0 pg, a net density of ~1400 kg/m3.
The mean atomic weight per atom in simple nanomechanical system
designs available in 2001 [192,
LINK]
ranged from 7.5-18.8 daltons/atom of structure, with an average of
12 daltons/atom; taking the average figure, the microbivore consists
of 610 billion structural or permanent atoms, plus ~15 billion
molecules of oxygen when fully charged at 1000 atm and 135 billion
molecules of water (solvating 2.52 billion glucose molecules) with
both chambers flooded. 4. Microbivore Performance and ApplicationsThis Section discusses the phagocytic activity of microbivores (Section 4.1), the pharmacokinetics of microbivores (Section 4.2), microbivore biocompatibility (Section 4.3), and various alternative applications for microbivores (Section 4.4).4.1 Phagocytic Activity of MicrobivoresTable 6 shows the approximate timeline for microbivore phagocytic activity during a single, complete microbe digestion cycle. One microbivore can completely digest one microbe that is up to ~2 microns3 in volume -- such as a P. aeruginosa bacterium -- in a time tdigest ~ 30 seconds. This is comparable to the 30-sec P. aeruginosa killing time of the chlorine dioxide/ammonia-based industrial chemical sterilant Cryocide [173] or the chemical germicide hydrogen peroxide [174], except that the microbivore also provides complete digestion of the pathogen. (Intravenous LD50 of H2O2 in rats is 21 mg/kg [175].) Larger microbes that are ~2-4 micron3 in volume could be completely internalized in ~2.5 seconds by taking two quick "bites," although full digestion requires two complete cycles or ~60 seconds, and still larger microbes could be ingested and digested piecemeal at a continuous rate of ~4 micron3/nanorobot-min, provided that some means can be found to avoid toxemia by ensuring that the watertight seal of a partially fragmented organism grappled against the nanorobot is maintained (possibly using flexible lipophilic flaps or metamorphic bumpers [1, LINK]). (Fungi are larger than bacteria but replicate more slowly and are less biotoxic, so the body's tolerance for material leakage during piecemeal ingestion of these organisms should be greater.) The microbivore consumes energy at a maximum rate of 200 pW, but more typically operates at ~100 pW.
Natural phagocytic cells are 100-1000 times larger in volume than microbivores but may consume almost as much power during comparable activities. For example, heat production rises from 9 pW in unstimulated human neutrophils up to 28 pW during phagocytosis, with the rise proportional to the number of particles ingested [176]. The basal rate for resting ~400 micron3 T-cell lymphocytes is ~20 pW, rising to ~65 pW during antigen response [177, 178]. Microbe ingestion times for natural professional phagocytes can be quite rapid, although complete digestion and excretion of the target pathogen may require hours. For example, 13.8-micron diameter murine bone-marrow macrophages have been observed ingesting a 15 micron particle in 30 minutes [179], whereas an ~8-micron lymphocyte was ingested by a macrophage in only 3 minutes with dramatic shape changes, including formation of a pseudopod 155 microns in length [180]. Nevertheless, while macrophages can ingest up to ~25% of their volume per hour [105], microbivores can process ~2000% of their volume per hour, thus are about 80 times more efficient as phagocytic agents, in terms of volume/sec digested per unit volume of phagocytic agent. Natural professional phagocytic cells such as neutrophils also have a maximum capacity for phagocytosis during their short lifetime, typically a few hours in blood or a few days in tissue. In one experiment [181], 1-100 S. aureus or S. faecalis bacteria were presented to each neutrophil (PMN), which digested more of them at the higher concentrations. At the highest concentration (100:1), PMNs from normal patients could only kill a mean of 9 S. aureus bacteria per PMN, while PMNs from carriers of of chronic granulomatous disease could kill a mean of 14 S. faecalis bacteria per PMN. By comparison, a single microbivore completely digests ~3000 microbes/day of P. aeruginosa bacteria with no well-defined maximum lifetime capacity for phagocytosis. 4.2 Microbivore PharmacokineticsTo crudely quantify the activity of a specific dose size of microbivores, a simple model of microbe-microbivore interaction may be constructed as follows.Consider a population of microbivores of spherical-equivalent radius RMV and number density nMV (nanorobots/m3), and a second population of microbes of spherical-equivalent radius Rbug and number density nbug (microbes/m3), simultaneously present in a fluid compartment of volume Vfluid, temperature Tfluid, and viscosity efluid. There are NMV = (nMVVfluid) microbivores and Nbug = (nbugVfluid) microbes initially present in the fluid compartment. After some incremental thermal diffusion time Dt each microbe migrates one diameter away from its previous position in the fluid. Any microbivore that is entirely present within a radius of (Rbug + 2RMV) of the center of the microbe's new position will be in collision with the microbe, hence the probability of collision is pcoll ~ (4/3) p nMV (Rbug + 2RMV)3 and the half-life for microbe-microbivore collision is t½ = Dt ln(½) / ln(1-pcoll) where Dt = 12 p efluidRbug3 / kT for an RMS displacement of one microbial diameter [1, LINK]. The half-life for microbe removal is therefore thalf = t½Ncoll, where Ncoll is the number of microbe-microbivore collisions required to ensure adhesion and capture. That is, after a time thalf has elapsed, the fixed population of microbivores has eliminated half of the original population of target microbes. This formulation assumes the usual therapeutic situation wherein a large surplus of nanorobots is present relative to the target microbes (NMV >> Nbug), in which case each microbivore only rarely consumes more than a single microbe during the therapeutic mission time tmission. This formulation allows us to ignore the microbivore phagocytic time tdigest ~ 30 sec (Section 4.1) as long as tmission > tdigest. However, microbes are not entirely passive targets for nanorobotic digestion. After one microbial replication time trepl has elapsed, all extant microbes produce a single daughter microbe, doubling the surviving population of microbes. The fastest known bacterial replicators have a mean generation time of 900-1200 sec [182, 183]. In one experiment, E. coli and P. aeruginosa replicating in the peritoneal cavities of mice having normal host clearance mechanisms displayed generation times of 33 min (1980 sec) and 20 min (1200 sec), respectively, during the first stages of infection [184]; in another experiment P. aeruginosa had a doubling time of 30-32 min (1800-1920 sec) while replicating in normal mouse lung but only 16 min (960 sec) in granulocytopenic (immune-compromised) mice [185]. (Enterobacteria such as E. coli divide only once every 12-24 hours when in the human colon (i.e., trepl = 43,200-86,400 sec) [186], far slower than the optimal laboratory batch rate of trepl ~ 1000 sec.) Using these relations and taking RMV = 1.42 microns, Rbug = 0.4 microns, efluid = 0.0011 kg/m-sec, k = 0.01381 zJ/molecule-K (Boltzmann's constant), T = 37°C, and trepl ~ 1000 sec, a mild bacteremia with nbug = 0.1 ×106 CFU/ml (Section 2.1.4) throughout a blood volume of Vfluid = 5400 cm3 is reduced from an initial bacterial load of Nbug = 5.4 ×108 CFU down to Nbug < 1 CFU in ~460 sec (~8 min) at Ncoll = 1 or in ~5400 sec (~1.5 hr) using a therapeutic dose of 1012 microbivores (a "terabot" dose). A severe bacteremia with nbug = 100 ×106 CFU/ml (Section 2.1.4) is eliminated in ~620 sec (~10 min) at Ncoll = 1 or in ~7300 sec (~2 hr) at Ncoll = 10. A single 1-terabot (1012-device) intravenous dose of microbivores constitutes a volume of ~12 cm3 of devices and produces a nanocrit of Nct ~ 0.2% when injected into a normal adult human male patient, and could liberate up to 200 watts of systemic waste heat which is very near the maximum thermogenic limit for in vivo medical nanorobot systems [1, LINK]. Similar bacteremias could be eliminated in 1.5 hr (mild case) to 2.1 hr (severe case) using a 0.1 terabot dose if Ncoll = 1, but the infection cannot be controlled with only 1011 microbivores if Ncoll = 10 because the bacteria can replicate faster than the fixed microbivore population can capture and digest them in this situation. The breakeven microbivore dose that is just large enough to prevent the microbial population from expanding, but is too small to reduce it, is obtained by setting trepl > ~ t½ and is given by:
While microbivores can fully eliminate septicemic infections in minutes to hours, natural phagocytic defenses -- even when aided by antibiotics -- can sometimes require weeks or months to achieve complete clearance of target bacteria from the bloodstream (Section 2.1). Thus microbivores appear to be up to ~1000 times faster-acting than either natural or antibiotic-assisted biological phagocytic defenses. Only when the pathogens are seriously crippled can the natural defenses achieve comparable clearance rates. For example, in one experiment [187] mice were able to clear ~80% of a 5000 CFU/gm dose of sialic acid-deficient group B streptococci by phagocytosis within 1 hour, whereas a like number of nondeficient streptococci similarly placed evaded phagocytic killing and disseminated to various tissues. Another useful comparative perspective is that the administration of antibacterial agents (e.g., against E. coli) typically may increase the LD50 of that pathogen by ~500-fold using antibiotics [30] or ~850-fold using monoclonal antibodies [188]. For example, the mammalian LD50 for E. coli is ~0.1-1 ×106 CFU/ml [27-30], rising to ~108 CFU/ml with the administration of antibiotics. By employing a suitable dose of microbivores, a bloodstream bacterial concentration up to the theoretical maximum of ~1011 CFU/ml (~20% of blood volume assuming ~2 micron3 organisms) could be controlled, bringing another ~1000-fold improvement using nanomedicine and at last extending the therapeutic competence of the physician to the entire range of potential bacterial threats, including locally dense infections. 4.3 Microbivore BiocompatibilityNanorobot biocompatibility [189-191] is a major topic whose complete discussion [192, LINK] is beyond the scope of this paper. A general observation is that it should be possible to endow nanorobots with surfaces of engineered nonadhesivity to serum opsonins and other bloodborne proteins [190], thus avoiding both nanorobot surface fouling and various systemic reactions such as complement activation, immune response, thrombogenicity, hypersensitivity, and nanopyrexia [191]. For example, biomimetic steric barriers (e.g., artificial glycocalyx [193]) might be deployed at the nanorobot surface as a coating over unoccupied hull areas of oligosaccharide surfactant polymers creating a 0.7-1.2 nm thick steric barrier at the nanorobot surface [160]. More research on devising such barriers in the nanomedical context is to be strongly encouraged.For microbivores, several additional biocompatibility issues also must be explicitly addressed. First, nanorobots larger than ~1 micron in all three physical dimensions are subject to possible geometrical trapping in the fenestral slits of the splenic sinusoids in the red pulp of the spleen [192]. A small percentage of blood is forced to circulate through a physical filter in the spleen requiring passage through slits measuring 1-2 microns in width and ~6 microns in length [194-196]. Microbivores which become pinned to a slit face-on, or which become stuck edge-on during an attempted passage, can detect that they have become trapped by measuring various blood component concentration and pressure differentials across their surfaces. The nanorobot then activates its automatic splenofenestral escape protocol, which involves the extension and patterned ciliation of surface grapples until sensor readings reveal that passage through the slit is complete, which is then followed by grapple retraction. Second, virtually every medical nanorobot placed inside the human body will encounter natural phagocytic cells many times during its mission [192, LINK]. Microbivores may incorporate any of several possible phagocyte avoidance and escape techniques [192, LINK], possibly including, for example, surface-tethered phagocyte chemorepellent molecules [197] or phagocyte engulfment inhibitors [198]. Third, the careless internalization of motile bacilli having long flagellar tails could result in the release of truncated bacterial tails into the bloodstream (Section 3.1.3). The typical bacterial flagellum is a close-packed rigid helix ~20 nm in diameter with a ~3 nm flagellin protein core, and its length is almost always >100 times its thickness [199], e.g., up to 10 microns long. There is significant antigenic diversity among bacterial flagellar epitopes [200-205] that white cells can recognize [206]. For example, Salmonella flagella are antigenically diverse and highly immunopotent [201] -- purified Salmonella typhi flagellar protein decreases CD14 expression and potently induces proinflammatory cytokine production (e.g., TNF-alpha, IL-6, IL-10, gamma interferon) by human peripheral blood mononuclear cells, and dramatically reduces expression of CD54 on macrophages, thus reducing the ability of those cells to take up soluble antigen [207]. Free releases of bacterial flagella into the bloodstream could produce inflammation or various immune system responses, thus should be avoided. Complete internalization of tail may be ensured by specialized operational routines (e.g., forced end-over-end rotation of an internalized microbe while inside the MC, thus completely spooling the tail into the microbivore before fully sealing the ingestion port door), by specialized mechanical tools or jigs (e.g., a counterrotating interdigitated-knobbed capstan-roller pair, not included in the present design), or by other means. The modulus of rigidity for representative Salmonella flagellum has been measured as ~1 ×1010 N/m2 [242]; from Eqn. 9.44 of Nanomedicine [1], the force required to buckle a 1-micron length of this flagellum is ~0.8 nN, far less than the ~100 nN force available from the MC chopping blade (Section 3.1.3). Another microbivore-specific biocompatibility issue derives from the onboard presence of active artificial digestive enzymes. Although occurrences should be rare, stray intact artificial enzymes could be missed by the extraction rotors or could suffer some form of partial degradation and subsequently be egested into the bloodstream. Such enzymes or enzyme fragments could exhibit immunogenic, inflammatory, or other harmful activity in the body [208-212], produce localized hyperenzymemia [213] (often itself benign, as in hypertransaminasemia [214, 215]), or could serve as unintentional inflammatory mediators [216]. Fortunately, these artificial enzymes should prove quite fragile outside of the relatively well-controlled and protective microbivore internal environment, and should be rapidly attacked by natural enzymes and quickly degraded to harmless peptides and amino acids. Given a proper enzyme-transport system design, the release rate of such molecules should be extremely low. Finally, the current microbivore design has an inherent minor iatrogenic vector vulnerability given that, in principle, an artificial virus could be created that would bind only to a region of the nanorobot surface that lies within the no-reach radius of the grapple arms. Since adjacent grapples cannot reach into this area either, a virus that affixes itself within the no-reach circle closest to the base of each grapple could not easily be dislodged mechanically. It may be possible to detect this unwanted passenger by noticing that some rotors are blocked in a particular area, but a forced reverse flow from blocked sorting rotors probably would not be sufficient to dislodge such a bound virion. In the case of a bloodborne virus, this is not a particularly serious concern since the virus was in the bloodstream anyway and little protection is conferred upon it simply by virtue of its being permanently bonded to the microbivore hull. The iatrogenic risk increases for more advanced microbivore-class nanorobots that can crawl through tissues, or move from organ to organ, or move between tissues and blood. This mobility creates a potential danger of inadvertently spreading a viral infection from one localized area to many other areas, should the virion subsequently become detached. For these devices, either an anti-blind-spot viral-removal protocol must be created and implemented, or else the blind spot must be removed by: (1) adding more angle links to the grapples, thus improving their reach; (2) positioning grapple silos closer together so an adjacent grapple arm can always reach into the blind spot; (3) adding specific virus dislodgement mechanisms analogous to physical wiper blades or localized jets of compressed gas at the base of every grapple silo; or (4) by otherwise eliminating the blind spot. 4.4 Extended ApplicationsThe present microbivore design has emphasized the phagocytosis of isolated bloodborne bacterial pathogens. But microbivores, as a general class of medical nanorobots, have much broader applicability which can only briefly be summarized here.4.4.1 Infections of Meninges and Cerebrospinal FluidMicrobivores could be useful in the treatment of infections of the meninges and the cerebrospinal fluid (CSF). For example, bacterial counts in the CSF of children [217] and rhesus monkeys [218] with Hemophilus influenzae meningitis can range from 102-109 CFU/ml, and 105-106 CFU/ml is sufficient to produce inflammation [219]. Rabbit models show that a single intravenous ampicillin dose of ~0.125 gm (0.8 mg/ml blood) reduces H. influenzae bacteria in CSF from 107 CFU/ml to 2.2 ×103 CFU/ml after 8 hours, a bacterial kill rate of4.4.2 Systemic Inflammatory Cytokine ManagementWith minor additions, microbivores could be used to combat toxemia, the distribution throughout the body of poisonous products of bacteria growing in a focal or local site, and other biochemical sequelae of sepsis. For instance, E. coli-induced septicemic shock in vervet monkeys occurred at 425 ×106 CFU/ml and LPS endotoxin rose from normal at 0.076 ng/ml to a maximum of 1.130 ng/ml blood concentration [222]. In another study, endotoxin levels during a gram-negative bacterial infection rose from 0.2 to 2 ng/ml in pig blood [223]. Eliminating a bloodstream concentration of ~2 ng/ml of ~8 kDa LPS endotoxin [224] would require the extraction and enzymatic digestion of ~8 × 1014 LPS molecules from the ~5400 cm3 human blood compartment, a mere ~800 LPS molecules per nanorobot assuming a single terabot dose (1012 devices) of modified microbivores.The high mortality associated with gram-negative sepsis is due in large measure to the patient's reaction to LPS, which induces the production of cytokines such as IL-1beta and IL-6 which leads to an uncontrolled inflammatory reaction resulting in tissue damage and organ failure [225]. Small quantities (~ng/ml) of LPS are released by living and growing bacteria (see previous paragraph), but the killing of bacteria using traditional antibiotic regimens often liberates large quantities of additional LPS, potentially up to ~105 ng/ml [225]. Such massive releases as occur with the use of antibiotics will not accompany the use of microbivores, because all bacterial components (including all cell-wall LPS) are internalized and fully digested into harmless nonantigenic molecules prior to discharge from the device. Microbivores thus represent a complete antimicrobial therapy without increasing the risk of sepsis or septic shock. (Note that while gram-positive organisms can also induce cytokine production, 100- to 1000-fold more gram-positive bacteria are needed to induce the same concentration of cytokines as are induced by gram-negative bacteria [225].) If the patient presents with a septic condition
before the microbivores are introduced, a substantial preexisting
concentration of inflammatory cytokines will likely be present and
must be extracted from the blood in concert with the principal
antibacterial microbivore treatment. All unwanted cytokine molecules
may be rapidly and systemically extracted from the blood using a
modest dose of respirocyte-class nanodevices [2,
LINK], a combination-treatment approach previously suggested
elsewhere [1,
LINK;
191,
LINK]. Specifically, a 1-terabot intravenous dose of micron-size
pharmacytes [1,
LINK,
LINK]
each having ~105 cytokine-specific molecular sorting
rotors and ~0.5 micron3 of onboard storage capacity could
reduce the blood concentration of ~20 kDa IL-1beta and IL-6
cytokines from LPS-elevated levels of ~100 ng/ml [225]
( A temporary sequestration of iron from the blood, mimicking the effect of lactoferrin released by natural phagocytes, could further enhance microbivore effectiveness by slowing the bacterial growth rate and increasing trepl. 4.4.3 Biofilm DigestionMicrobivores, slightly altered, could also be used to digest bacterial biofilms [227]. Biofilms may vary widely in thickness, which is limited more by nutrient transport than by surface roughness. In vitro experiments show that aerobic Pseudomonas aeruginosa biofilms can grow to 30-40 microns in depth as monocultures, but may increase in depth to 130 microns when the culture is amended with anaerobic bacteria [228]. Microbivores can digest biomaterial at a rate of ~4 micron3/min, hence an array of closely packed microbivores (~6.8 micron2/device) attached to a biofilm can consume the biofilm at a rate of ~10 nm/sec, requiring ~105 sec (~3 hr) to consume an entire 100-micron thick biofilm. Again, some means must be found to ensure a watertight seal between partially fragmented organisms and the microbivore ingestion port (Section 4.1).4.4.4 Bacterial Infections in Other Fluids and TissuesBacteria present in sputum or in the mucous layers of the throat may be pursued by somewhat larger ambulatory microbivores having an additional array of longer grapples that could serve as locomotive mechanisms (legs), thus permitting the nanorobots to engage in microbial search-and-destroy missions along the luminal surfaces of the human trachea, bronchi, and bronchioles [229]. Normally there may be ~105 CFU/ml bacteria colonizing the oropharynx [230], >107 CFU/ml in sputum or throat swab during respiratory infections [231] or cystic fibrosis [232], and sputum infections up to ~4 ×108 CFU/ml have been reported [233, 234].With additional modifications, other variants of microbivores could patrol tissues, organs, and nonsanguinous fluid spaces such as pleural [235], synovial [236], or urinary fluids (e.g., asymptomatic bacteriuria has >105 CFU/ml urine [237], >103 CFU/ml is pathogenic [238] or >102 CFU/ml with dysuria [237]), pursuing bacteria as they disseminate beyond the bloodstream. Vasculomobile microbivores could follow cytokine gradients and collect at sites of infection, thus increasing their microbicidal efficiency. 4.4.5 Viral, Fungal, and Parasitic InfectionsMicrobivores can rid the blood of viral pathogens, which are typically present during viremia at concentrations similar to those found in bacteremia, ~0.1-100 ×106/ml (Section 2.2). Viruses tend to be much smaller than most bacteria, so processing time per virion may be considerably reduced, perhaps 5-10 seconds or less. Apparently the human body is already fairly efficient at removing virus particles from the bloodstream -- for instance, in one study of HIV-1 infected patients, measurements of plasma virus loads found that individual virions had a clearance half-life of 28-100 min for HIV-1 and 100-182 min for hepatitis C (HCV) virus [239]. The difficulty for the natural defensive systems is that replacement viruses are rapidly replicated and discharged into the blood by infected cells, thus perpetuating the infection. For example, the daily particle production rate in HIV-1 infected patients has been estimated as 2-16 ×109 particles/day for HIV-1 and 0.4-10 ×1012 particles/day for HCV [239]. Such production rates are nevertheless easily controlled by a terabot population of microbivores which has a collective digestive capacity of >1015 virions/day. One additional complication, well within the competence of the the current microbivore design, is that some viruses like HIV are mutating constantly, so that one patient may have as many as 8-10 different strains concurrently, all of which must be successfully recognized and eliminated.Fungemias involving particle loads of 1-1000 CFU/ml (Section 2.3) are rapidly cleared by microbivores. Fungal particles may be up to ~400 micron3 in volume, requiring ~100 min for complete digestion using a microbivorous protocol that employs careful piecewise digestion involving ~800 "bites" (Section 4.1). Blood parasites of comparable size (Section 2.4) may be present at concentrations similar to those found in bacteremia but may be controlled with terabot doses of microbivores. 4.4.6 Other ApplicationsMicrobivores could be designed to trap and retain (without digesting) samples of unknown microbes found floating in the bloodstream, when those microbes fall within a certain physician-specified size range and are confirmed not to be platelets or chylomicrons. These samples could then be returned to the attending physician for further investigation, following nanapheresis. Ranging still further afield, microbivore-derived devices could be employed in veterinary and military applications; to disinfect surfaces, objects, and volumes (e.g., 102-105 CFU/ml bacteria found in the sink fluid of washbasin drains in a pediatric ward [240]) or to sterilize organic samples or edible foodstuffs; to clean up biohazards, biopolluted drinking water, toxic biochemicals, or other environmental organic materials spills, as in bioremediation; and in many other useful applications.5. ConclusionsThis paper presents a theoretical nanorobot scaling study for artificial mechanical phagocytes of microscopic size, called "microbivores," whose primary function is to destroy microbiological pathogens found in the human bloodstream using a digest and discharge protocol. Some images of microbivores are now available online.The microbivore is an oblate spheroidal nanomedical device measuring 3.4 microns in diameter along its major axis and 2.00 microns in diameter along its minor axis, consisting of 610 billion precisely arranged structural atoms in a gross geometric volume of 12.1 micron3. During each cycle of operation, the target bacterium is bound to the surface of the microbivore via species-specific reversible binding sites. Telescoping robotic grapples emerge from silos in the device surface, establish secure anchorage to the microbe's plasma membrane, then transport the pathogen to the ingestion port at the front of the device where the cell is internalized into a morcellation chamber. After sufficient mechanical mincing, the morcellated remains are pistoned into a digestion chamber where a preprogrammed sequence of engineered enzymes are successively injected and extracted, reducing the morcellate primarily to monoresidue amino acids, mononucleotides, glycerol, free fatty acids and simple sugars, which are then harmlessly discharged into the environment, completing the cycle. The device may consume up to 200 pW of continuous
power while completely digesting trapped microbes at a maximum
throughput of 2 micron3 of organic material per 30-second
cycle. Microbivores are up to ~1000 times faster-acting than either
natural or antibiotic-assisted biological phagocytic defenses, and
are ~80 times more efficient as phagocytic agents than macrophages,
in terms of volume/sec digested per unit volume of phagocytic agent.
Besides intravenous bacterial scavenging, microbivores or related
devices may also be used to help clear respiratory, urinary, or
cerebrospinal bacterial infections; eliminate bacterial toxemias and
biofilms; eradicate viral, fungal, and parasitic infections;
disinfect surfaces, foodstuffs, or organic samples; and help clean
up biohazards and toxic chemicals. 6. AcknowledgementsThe author thanks Stephen S. Flitman, M.D., C. Christopher Hook, M.D., Ronald G. Landes, M.D., and also Forrest Bishop, Robert J. Bradbury, and Ralph C. Merkle, for helpful comments on an earlier version of this paper; Forrest Bishop for 3D modeling sutdies; and Robert J. Bradbury for preparing the hypertext version of this document.7. References
Copyright © 2001-2004 Robert A. Freitas Jr. All Rights Reserved | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
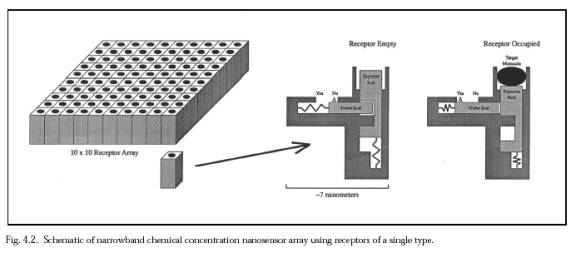{kind=link}